





.png)


















2021-12-31
導讀
串聯質譜(MS/MS)技術已經廣泛應用于新生(shēng)兒遺傳代謝病篩查項目,但存在假陽性率較高的問題。根據大(dà)樣本彙總分(fēn)析的結果,我(wǒ)國串聯質譜技術用于新生(shēng)兒遺傳代謝病篩查的初篩陽性率約爲1.91%,而最終确診陽性率僅爲0.04%,即每1萬例新生(shēng)兒中(zhōng),約有191例初篩爲陽性,經召回複查及進一(yī)步檢測,最終僅有4例确診。造成串聯質譜篩查假陽性結果的因素有很多,本期我(wǒ)們探讨攜帶者狀态對于篩查結果的影響。
什麽是攜帶者狀态
絕大(dà)多數遺傳代謝病爲常染色體(tǐ)隐性遺傳模式,即隻有當同源染色體(tǐ)上的兩份基因拷貝均發生(shēng)變異時才會導緻疾病的發生(shēng),而僅有1份拷貝發生(shēng)變異不會緻病,此時稱之爲攜帶者(carrier)或攜帶者狀态(carrier status)。
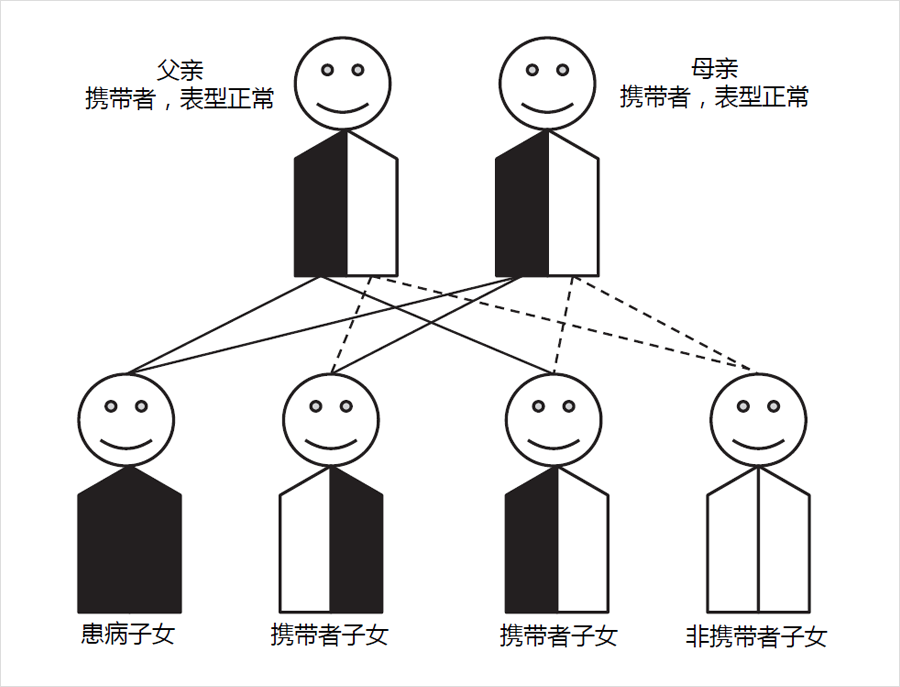
攜帶者狀态對新生(shēng)兒串聯質譜篩查結果的影響
由于正常等位基因的劑量補償效應,遺傳代謝病基因變異攜帶者不會患病,但與完全正常人群相比,其酶活性或多或少會受到一(yī)定程度的影響。對于新生(shēng)兒遺傳代謝病串聯質譜篩查,這種影響可能會體(tǐ)現在特定疾病相應指标出現異常,尤其是對于那些指标特異性較差的疾病,如中(zhōng)鏈酰基輔酶A脫氫酶缺乏症(MCADD)和極長鏈酰基輔酶A脫氫酶缺乏症(VLCADD)等。
01 中(zhōng)鏈酰基輔酶A脫氫酶缺乏症(MCADD)
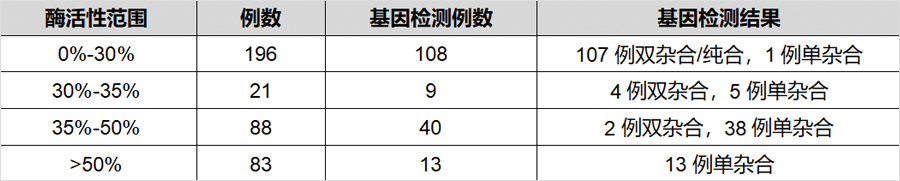
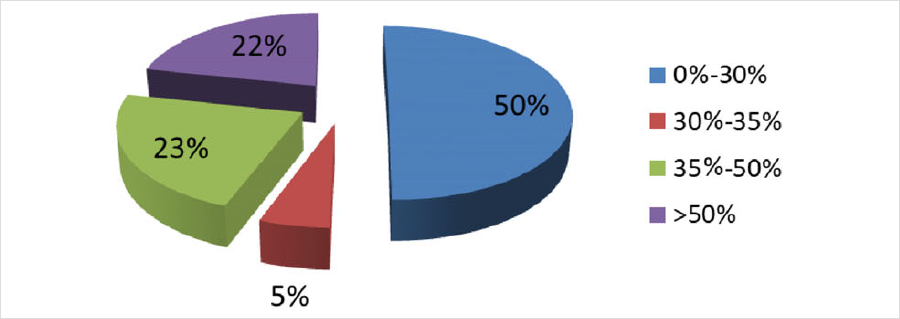
發表在《J Inherit Metab Dis》雜(zá)志(zhì),題爲“Genotype and residual enzyme activity in medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: Are predictions possible?”的研究[1],報道了460例串聯質譜篩查疑似MCADD的新生(shēng)兒,通過酶活性檢測和/或基因檢測進行确診,并分(fēn)析了基因型和酶活性的相關性。在388例進行了酶活性檢測的新生(shēng)兒中(zhōng),196例酶活性介于0%-30%(疑似患者),21例酶活性介于30%-35%(疑似患者或攜帶者),88例酶活性介于35%-50%(疑似攜帶者),其餘酶活性>50%(疑似正常)。171例新生(shēng)兒同時進行了基因檢測(44%),确診57例爲攜帶者。粗略推算因攜帶者狀态導緻的假陽性比例在40%以上。
02 極長鏈酰基輔酶A脫氫酶缺乏症(VLCADD)
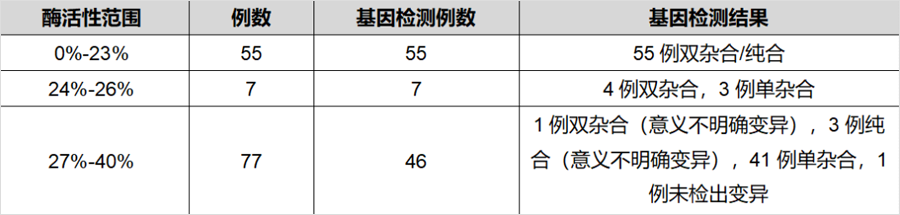
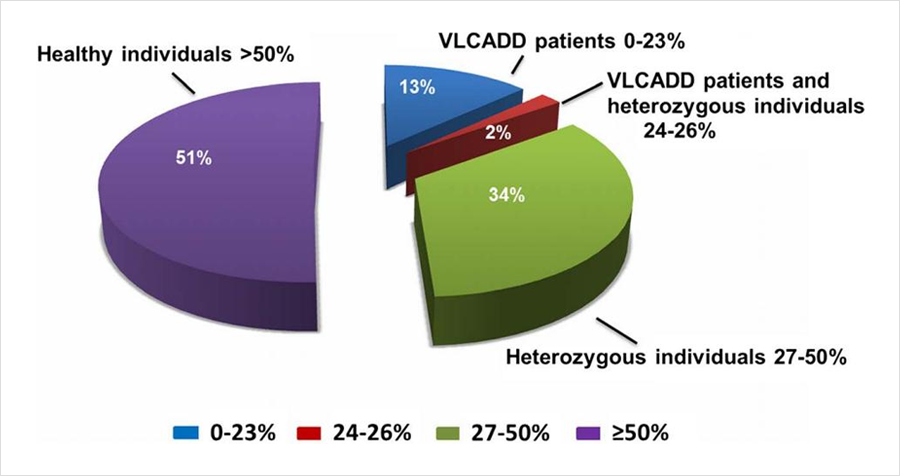
同樣發表在《J Inherit Metab Dis》雜(zá)志(zhì),題爲“The diagnostic challenge in very-long chain acyl-CoA dehydrogenase deficiency (VLCADD)”的研究[2],報道了403例串聯質譜篩查疑似VLCADD的新生(shēng)兒,采用酶活性檢測和/或基因檢測進行确診。55例酶活性介于0%-23%(疑似患者),7例酶活性介于24%-26%(疑似患者或攜帶者),138例酶活性介于27%-50%(疑似攜帶者),203例酶活性>50%(疑似正常)。108例酶活性<40%的新生(shēng)兒同時進行了基因檢測,确診44例爲攜帶者。粗略推算因攜帶者狀态導緻的假陽性比例接近40%。
03 血酰基肉堿水平是否可提示攜帶者狀态?
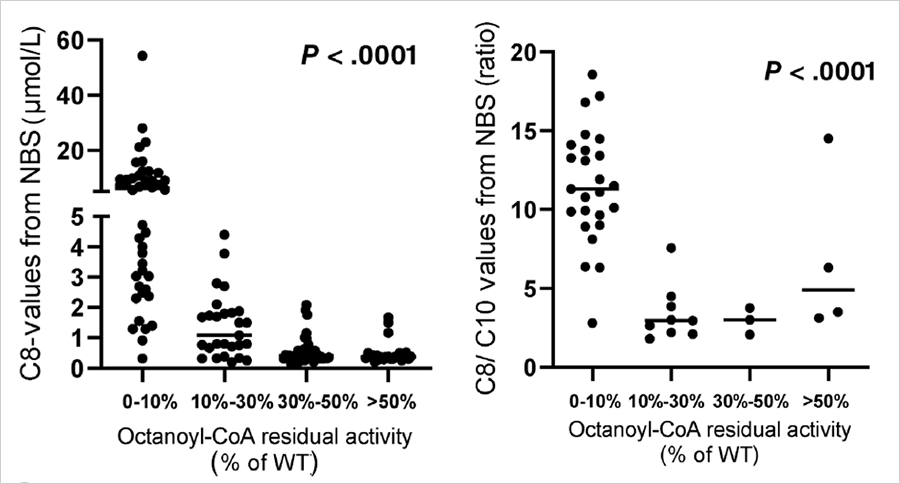
血辛酰肉堿(C8)及C8/C10比值增高是串聯質譜篩查MCADD的主要診斷指标,那這兩個指标是否可用來初步區分(fēn)患者與攜帶者呢?在上述的MCADD研究中(zhōng),研究人員(yuán)做了酶活性與各個指标相關性的分(fēn)析。如下(xià)圖所示,酶活性介于0%-10%的MCADD患者,其串聯質譜C8和C8/C10值均顯著高于其他個體(tǐ),但存在一(yī)定程度的交叉;而酶活性介于10%-30%的患者,與攜帶者和正常個體(tǐ)相比,沒有顯著差異。因此,通過C8和C8/C10數值進行患者和攜帶者的區分(fēn)是不可行的。
思考與總結
攜帶者狀态是導緻新生(shēng)兒串聯質譜MCADD和VLCADD篩查假陽性結果的主要原因之一(yī)。由于酶活性檢測的普及率較低,基因檢測是目前确診的主要手段。但彙總既往研究報道[3-5],我(wǒ)國MCADD患兒基因診斷的靈敏度僅爲78.9%(彙總分(fēn)析的38例經臨床、血生(shēng)化、血肉堿或尿有機酸檢測等确診的患兒中(zhōng),8例僅檢測到單雜(zá)合變異),這與本文提到的國外(wài)研究存在較大(dà)差異。因此,在應用基因檢測技術對串聯質譜篩查陽性的MCADD患兒進行輔助診斷時,如果僅檢出單雜(zá)合變異,除考慮攜帶者狀态造成的假陽性可能外(wài),也不能排除基因檢測假陰性的可能。而對于VLCADD,我(wǒ)國患兒基因診斷的靈敏度接近100%[6-8],因此,檢出單雜(zá)合變異時不需要考慮假陰性的可能。
參考文獻:
1. Tucci S, Wagner C, Grünert SC, et al. Genotype and residual enzyme activity in medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: Are predictions possible?. J Inherit Metab Dis. 2021;44(4):916-925.
2. Hesse J, Braun C, Behringer S, Matysiak U, Spiekerkoetter U, Tucci S. The diagnostic challenge in very-long chain acyl-CoA dehydrogenase deficiency (VLCADD). J Inherit Metab Dis. 2018;41(6):1169-1178.
3. Gong Z, Liang L, Qiu W, et al. Clinical, Biochemical, and Molecular Analyses of Medium-Chain Acyl-CoA Dehydrogenase Deficiency in Chinese Patients. Front Genet. 2021;12:577046.
4. Li Y, Zhu R, Liu Y, Song J, Xu J, Yang Y. Medium-chain acyl-coenzyme A dehydrogenase deficiency: Six cases in the Chinese population. Pediatr Int. 2019;61(6):551-557.
5. 童凡, 蔣萍萍, 楊茹萊,等. 中(zhōng)鏈酰基輔酶A脫氫酶缺乏症新生(shēng)兒篩查及随訪研究[J]. 中(zhōng)國當代兒科雜(zá)志(zhì), 2019, 21(1):6.
6. 曹金俊, 邱文娟, 章瑞南(nán),等. 極長鏈酰基輔酶A脫氫酶缺乏症11例的臨床和ACADVL基因突變譜分(fēn)析[J]. 中(zhōng)華兒科雜(zá)志(zhì), 2015(4):6.
7. Li X, Ma R, Liu Y, et al. One potential hotspot ACADVL mutation in Chinese patients with very-long-chain acyl-coenzyme A dehydrogenase deficiency. Clin Chim Acta. 2020;503:218-222.
8. 錢古柃,洪芳,童凡,等. 極長鏈酰基輔酶A脫氫酶缺乏症16例臨床及基因型分(fēn)析[J]. 兒科藥學雜(zá)志(zhì), 2020, 26(9):4.
































